


AKV9, Protein Aggregation Inhibitor
AKV9 is the first compound reported to improve the health of diseased upper motor neurons by:
Although different proteins aggregate in different neurons and diseases, the underlying causes of neuronal pathology appear to be shared. Therefore, developing therapies that target protein aggregation would have broad implications.
In ALS, misfolded mutant superoxide dismutase 1 (SOD1) proteins aggregate in diseased motor neurons, and this accounts for about 10% of familial ALS. In addition, TDP-43 pathology, which is characterized by the accumulation of aggregated proteins that contain phosphorylated TDP-43 in the cytoplasm, accounts for more than 90% of all ALS cases. Since misfolded SOD1 toxicity and TDP-43 pathology do not overlap, inhibitors that act on both of these protein aggregates offer broad implications for a larger population of ALS.
Therefore, we investigated compounds for their ability to reduce protein aggregation.
With funding from the ALS Association, Department of Defense, and National Institutes of Health, a library of >50,000 drug-like compounds was screened to identify hits for inhibition of protein aggregation and toxicity in cells expressing the mutant SOD1 gene. Of these, 68 active hits were identified using a 60% efficacy cutoff.
Computational analysis identified 17 distinct chemical scaffolds containing structurally related active and inactive compounds plus 15 singleton hits. Compounds or series were deprioritized or deselected from review when structurally similar compounds were inactive, a low number of active analogues were found, toxicity liabilities were identified, no preliminary structure activity relationship (SAR) could be established, there was a synthetic challenge to make analogues rapidly, or based on a medicinal chemistry review.
At the end of this iterative process, three distinct and validated chemical series were selected for medicinal chemistry optimization. Several years of optimization of the active scaffolds was carried out, resulting in four optimized compounds, one of which was AKV9. These compounds were selected based on their ability to inhibit protein aggregation and toxicity, as well as having excellent in vitro/in vivo pharmacokinetic properties.
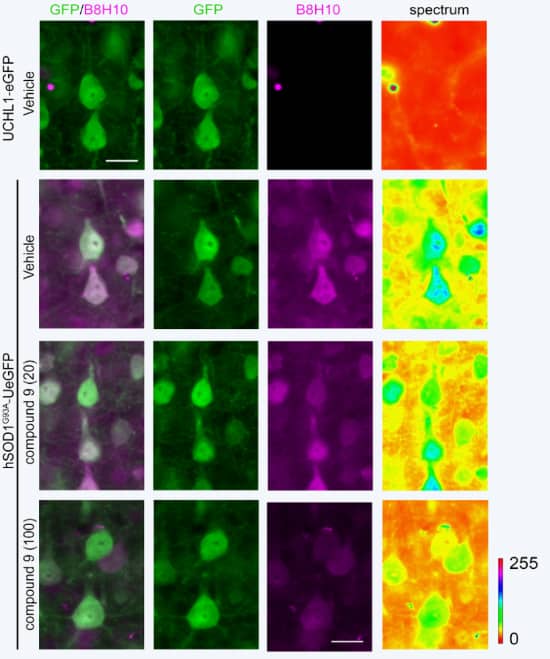
AKV9 was identified on the basis of its ability to reduce aggregated mSOD1 in PC12 cells.
Here, aggregated mSOD1 is shown in UMNs of an ALS mouse model. B8H10 is a monoclonal antibody that can detect aggregated SOD1 mutants but not normal SOD1. Healthy mice (3rd column, top box) show no mutant SOD1. Mice with mutant SOD1 show much aggregation (3rd column, 2nd box). When treated with AKV9 with 20 mg/kg/day for 60 days, much less aggregated mutant SOD1 occurs (3rd column, 3rd box), and when treated with AKV9 at 100 mg/kg/day for 60 days, very little aggregated mutant SOD1 is detected (3rd column, 4th box).
There is an urgent need to develop preclinical assays that include UMN health as a readout. A novel in vitro platform was developed by SAB member, Dr. P. Hande Ozdinler, using an UMN reporter line in which UMNs having misfolded SOD 1 toxicity, were genetically labeled with fluorescence. AKV9 (aka, NU-9), edavarone, and riluzole were independently tested in this model and also AKV9 in combination with either edavarone or riluzole. AKV9 was found to be more effective at enhancing axon length than either edavarone or riluzole and when used in combination, had an additive effect on axon length with more enhanced branching and arborization, two important characteristics of healthy UMN in vitro.

G SFM, healthy mice;
GS SFM, mutant SOD1 mice;
GS NU-9, mutant SOD1 mice treated with NU-9;
GS Riluzole, mutant SOD1 mice treated with riluzole;
GS NU-9 + Riluzole, mutant SOD1 mice treated with NU-9 and riluzole;
GS Edaravone, mutant SOD1 mice treated with edaravone;
GS NU-9 + Edaravone, mutant SOD1 mice treated with NU-9 and edaravone

Apical dendrites are the site of cortical integration, and their stability is important for proper upper motor neuron (UMN) modulation, function, and health. Improving cytoarchitectural integrity of UMN apical dendrites by reducing vacuolization and disintegration is imperative. Unlike untreated mutant SOD1 mice (middle), which have vacuoles and disintegration (arrows), mutant SOD1 mice treated with AKV9 (right) have apical dendrites as healthy as those of healthy mice (left).
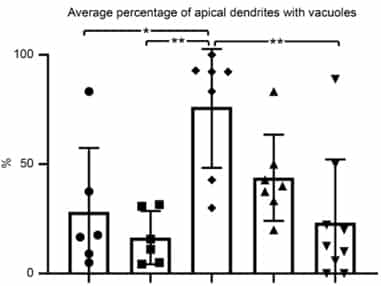
The percentage of upper motor neurons (UMNs) with vacuoles in healthy mice (left two bars: left one untreated, right one with AKV9) is relatively low (about 25%).
For UMNs that are diseased with mSOD1 toxicity (middle bar), there is a high percentage of vacuoles.
When mutant SOD1 mice are treated with 20 mg/kg/day for 60 days (2nd bar from right) and 100 mg/kg/day for 60 days (right-hand bar), vacuolization is low, and at 100 mg/kg/day for 60 days UMNs of diseased mice become comparable to the healthy UMNs in control group (compare last and first bar graphs)
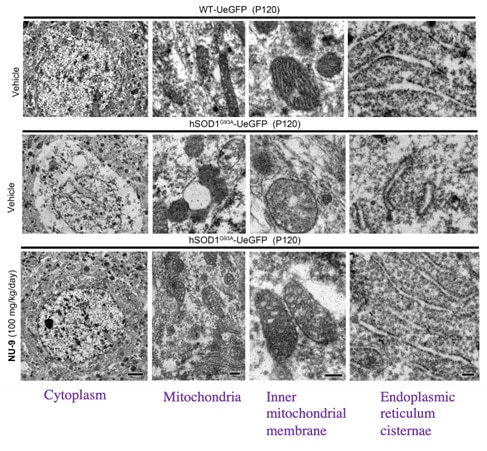
The mitochondria, inner mitochondrial membrane and endoplasmic reticulum of upper motor neurons (UMNs) from mice with mutant SOD1 treated with 100 mg/kg/day for 60 days AKV9 (bottom row of electron microscopy images) look like those from healthy mice (upper row of images), but the untreated mice with mutant SOD1 (middle row of images) are degenerated.
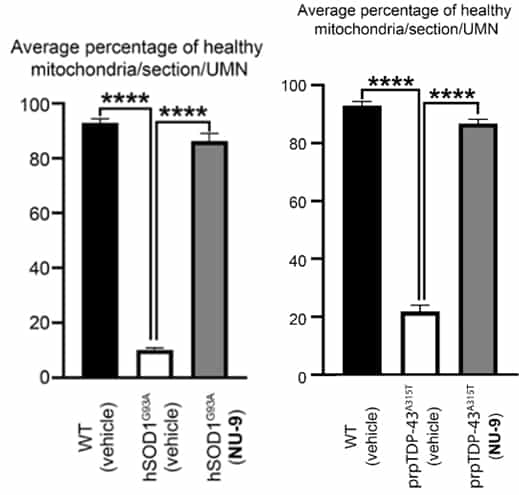
Upper motor neurons (UMNs) of mutant SOD1 (left figure) and mutant TDP-43 mice (right figure) experience degeneration of their organelles in the UMN soma (middle bar left figure for mice with mutant SOD1 and middle bar right figure for mice with mutant TDP-43, right figure).
Treatment of the mice with AKV9 (aka, NU-9) with 100 mg/kg/day for 60 days (right bar in each graph) resulted in mitochondria that are as healthy as the mitochondria that are found in the UMNs of control mice (WT, left bar in each graph).

There is a dose-dependent increase in upper motor neuron (UMN) health starting at P60, when symptoms appear and when AKV9 dosing starts. Healthy mice (left bar) have a large number of healthy UMNs. Mutant SOD1 mice (2nd bar) have few healthy UMNs. When treated with AKV9 (aka, C9) at 20 mg/kg/day for 60 days (3rd bar), there is an increase in the number of healthy UMNs, and when treated with AKV9 at 100 mg/kg/day for 60 days (4th bar), there are almost an equivalent number of healthy UMNs to wild-type, healthy mice. A similar result was obtained with mutant TDP-43 mice.
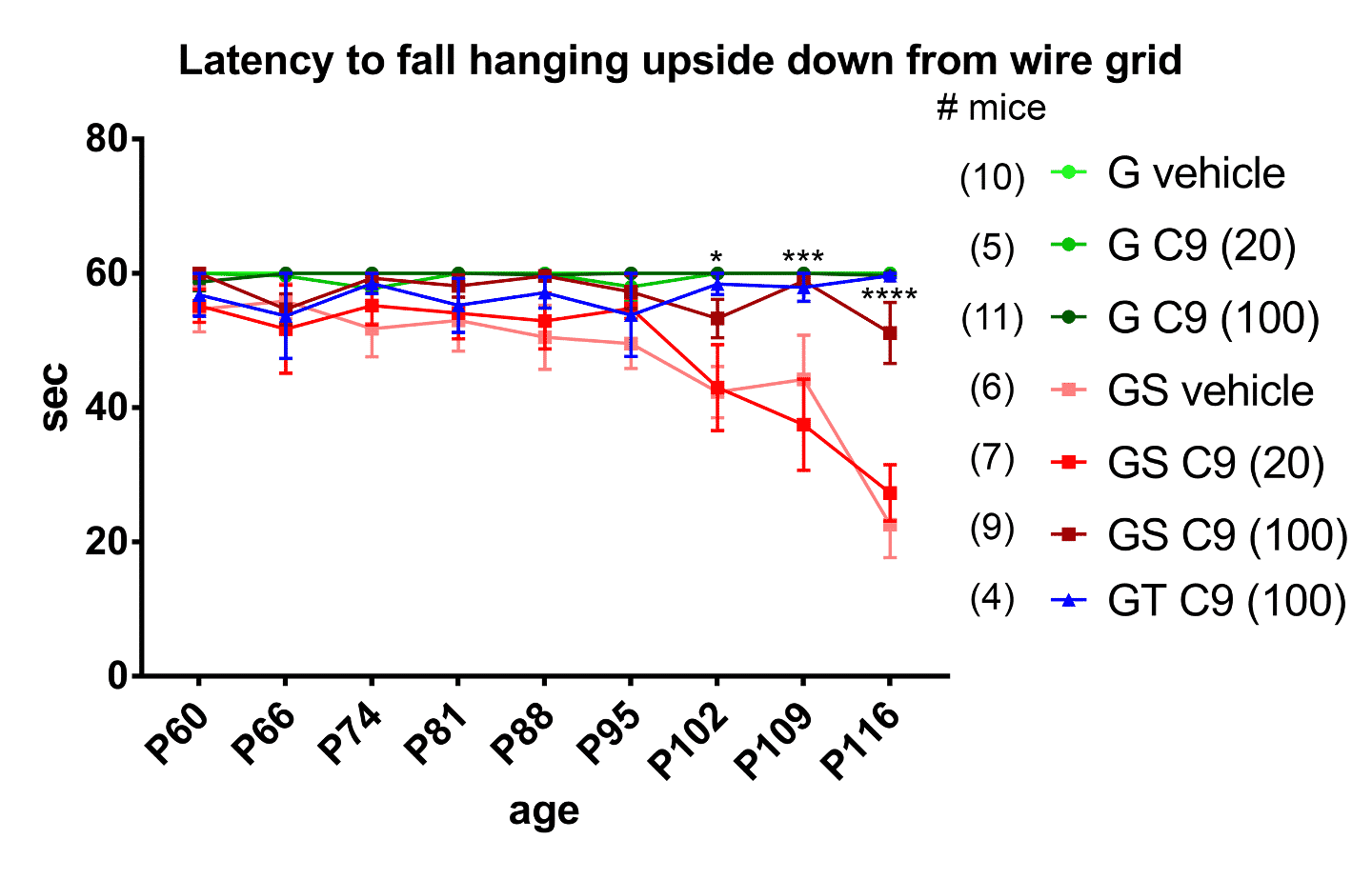
The inverted wire mesh test, in which the mouse has to hold on to an inverted mesh, is directed at UMN health and muscle strength. About 95 days after birth (P95) the mutant SOD1 mice (GS vehicle) start losing their grip and fall off sooner than mutant SOD1 or mutant TDP-43 mice (GT) treated with AKV9 at 100 mg/kg/day for 60 days, which hold on as long as the healthy mice (all of the G mice).
Please refer to the manuscript for details.
These results, obtained from two different mouse models of ALS that represent two independent underlying causes of ALS (i.e., misfolded SOD1 toxicity and TDP-43 pathology), are the first evidence that reveals the impact of AKV9 (aka, C9) for improving the health of diseased UMNs and ultimately, effective treatments for patients.
What Makes AKV9 Different?
Previous ALS drug discovery efforts
- Mostly focused on improving the life extension of ALS mouse models to suggest improved patient outcome, believing that mice mimic humans
- Never considered improving the health of diseased UMNs as a potential treatment strategy
- At times utilized cells and cell lines that are not related to motor neurons to obtain data about motor neuron health
- Could not show translation between mouse survival and human survival
AKAVA’s unique approach
- Utilizes a cell-based and mechanism-focused drug discovery effort
- Focuses on the biology and pathology of diseased upper motor neurons (UMNs) with the goal of improving their health at a cellular level
- Investigates and obtains data directly from diseased UMNs and not from unrelated cell models
- Builds on the fact that translation is not at a species level, but is at a cellular level
Ornithine Aminotransferase Inactivators
The gene that encodes the enzyme ornithine aminotransferase (OAT) is overexpressed in rats with hepatocellular carcinoma (HCC). Development of HCC correlates with activation of OAT.
We have shown that inhibition of OAT leads to inhibition of HCC growth.
Our inactivators of OAT inhibit secretion of alpha-fetoprotein, a biomarker for HCC, and tumor growth in mice with human-derived HCC. Five different molecules are undergoing comparative efficacy and toxicology studies to determine which to move into IND-enabling studies.

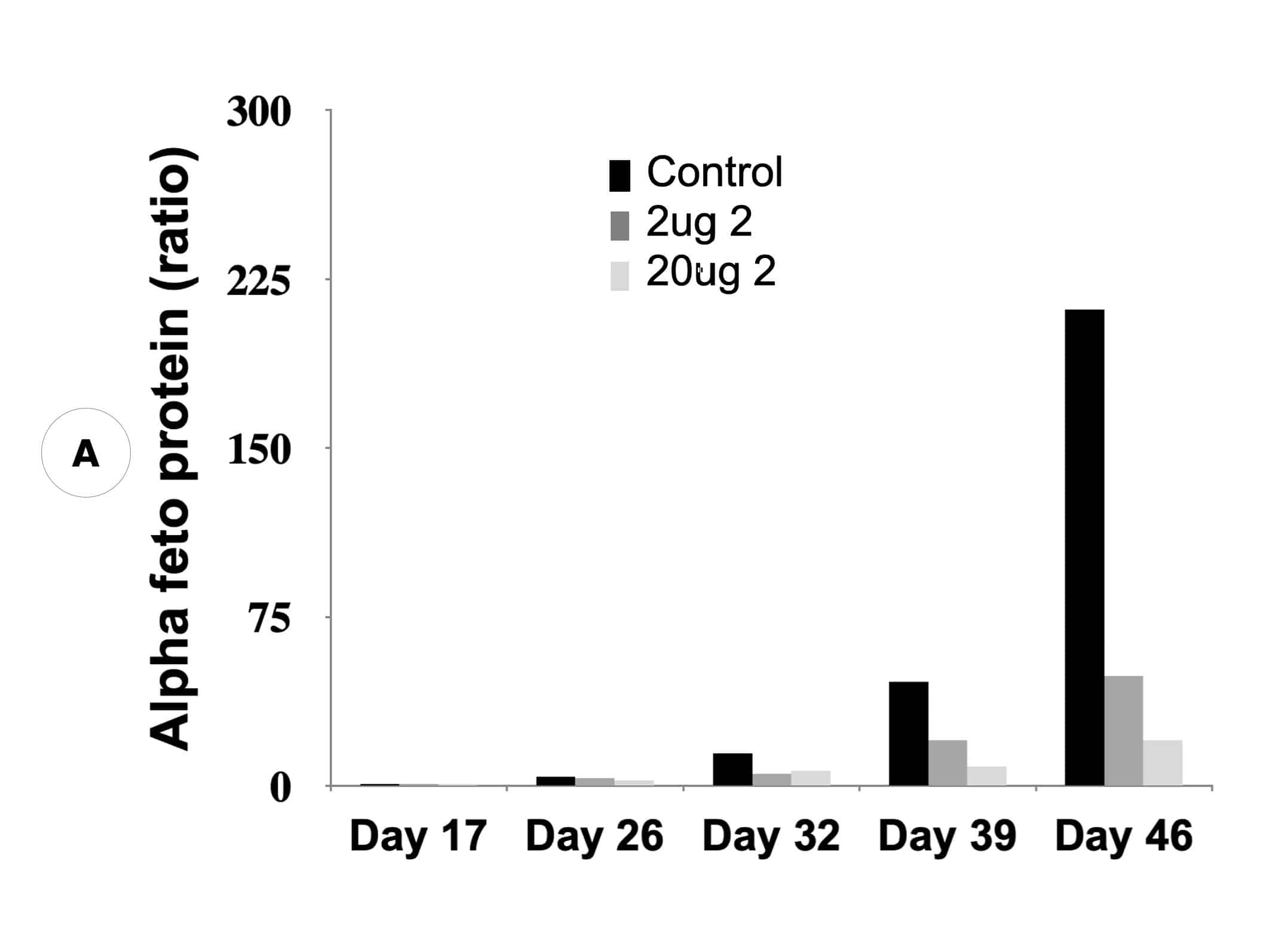
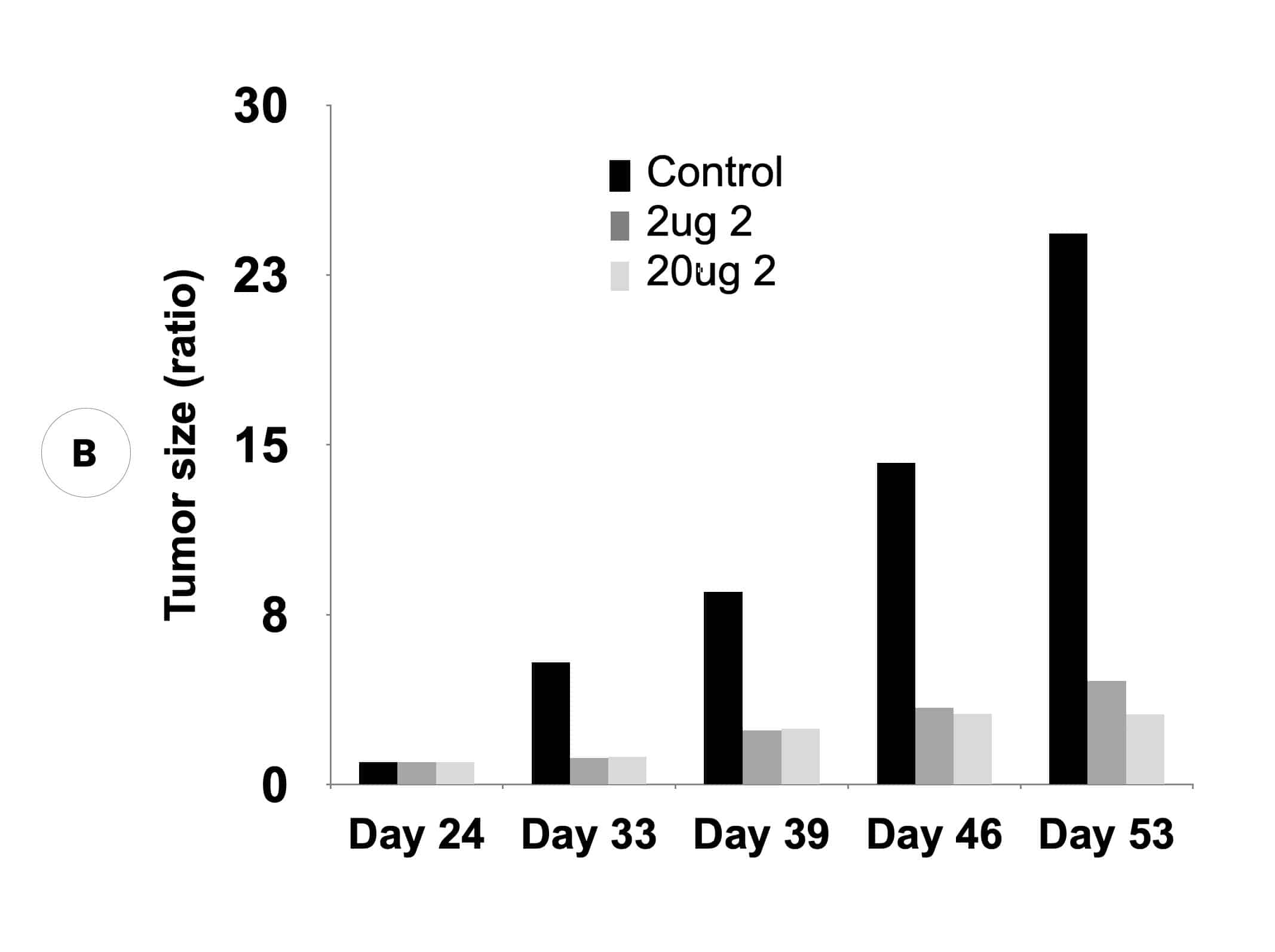
Suppression of biomarker, alpha-fetoprotein, and HCC tumor size in nude mice. (A) Compound 2 administered at either 2 micrograms (0.1 mg/kg) or 20 micrograms (1 mg/kg) per mouse and biomarker secretion determined. (B) Inhibition of tumor growth at the same concentrations correlates with inhibition of the biomarker.
Hepatocellular carcinoma (HCC) is the most common primary liver cancer and the 3rd leading cause of cancer death worldwide; over 800,000 people worldwide contract HCC and about 700,000 deaths occur annually.
Treatments include:
- surgery
- radiation therapy
- chemotherapy
- immunotherapy
- targeted therapy
- hormone therapy
- stem cell transplant
- precision medicine
Despite all of the approaches available, the 5-year survival rate is only 10%, and side effect profiles are high.
The development of HCC has been shown to correlate with the activation of the Wnt/β-catenin signaling pathway in liver. The Wnt/β-catenin pathway, an evolutionarily conserved pathway, is essential to normal cellular processes such as growth, development, survival, and regeneration.
β-Catenin serves three major roles in liver physiology. In the presence of Wnt, β-catenin translocates to the nucleus, where it functions to activate genes essential for proliferation, growth, and regeneration of the liver. However, in addition to its diverse important physiological functions in liver, β-catenin also is associated with the initiation and progression of liver cancer, generally resulting from mutations in members of the Wnt/β-catenin pathway.
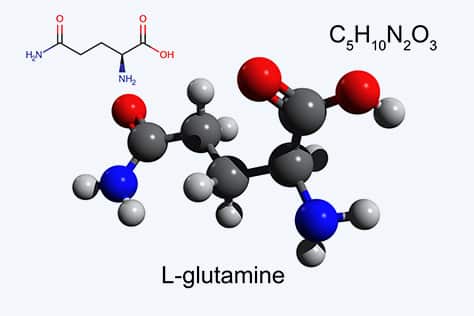
Activation of the Wnt/β-catenin signaling pathway, and concomitant development in liver of HCC, correlates with the upregulation of Wnt/β-catenin pathway proteins ornithine aminotransferase (OAT), glutamate transporter GLT-1, and glutamine synthetase; loss of β-catenin induction of those three proteins blocks glutamine synthesis as a result of loss of OAT activity. L-Glutamine is essential for growth of both normal and neoplastic cells; however, tumor cells take up glutamine more efficiently than normal cells, and tumor growth is enhanced by glutamine.
Cancer cells distinguish themselves from normal cells in that they have an increased requirement for glutamine to support anabolic processes that stimulate proliferation. Increased activity of OAT by Wnt/β-catenin activation enhances tumor cell growth, allowing a controlled growth advantage to the tumor cell. Therefore, reducing enhanced glutamine concentrations by OAT inhibition should inhibit tumor growth and be a viable antitumor mechanism, and we have found that inhibition of OAT does inhibit the growth of HCC in mice.
Neuronal Nitric Oxide Synthase Inhibitors
Neuronal nitric oxide synthase (nNOS) is an enzyme in neuronal cells that catalyzes the production of the cell-signaling molecule nitric oxide, which is a neurotransmitter important for brain development, memory, and learning. However, pathological overproduction of nitric oxide, which is a free radical, is detrimental to neuronal tissue and can lead to all of the neurodegenerative diseases.
Melanocytes, melanin-producing cells in the skin, are derived from the neural crest and have been shown to contain nNOS. UV radiation causes an upregulation of nNOS, which increases the production of nitric oxide that can degenerate the melanocytes, leading to melanoma.
Therefore, the connection between neurodegeneration and melanoma is formation of excess nitric oxide. Inhibitors of nNOS reduce the production of nitric oxide and could have a therapeutic effect against neurodegeneration and melanoma.

Neurodegeneration
Our selective neuronal nitric oxide synthase (nNOS) inhibitors prevent the phenotype of cerebral palsy in rabbits.
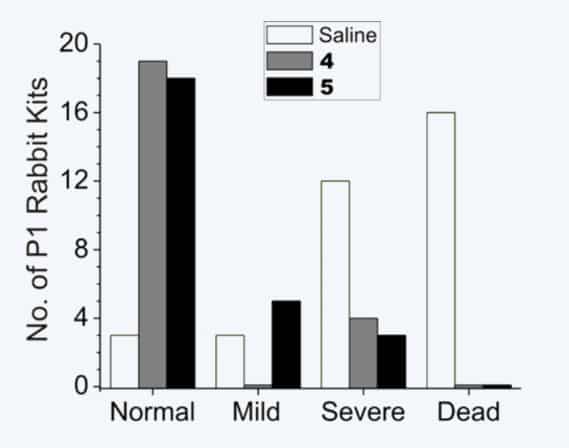
Neurobehavioral effects of two nNOS-selective inhibitors (4 and 5) on rabbit kits exposed to hypoxia-ischemia. Blockage of oxygen to fetuses during pregnancy is an important cause for cerebral palsy in humans. 16/34 (47%) of rabbit kits from saline-treated dams died, but no deaths (0/49) were observed from treated animals. Of the kits from saline-treated dams that came to term (18/34), severe neurobehavioral abnormalities occurred in 12/18 (67%) compared to only 7/49 (14%) in dams treated with 4 or 5. Furthermore, the 4 or 5-treated animals exhibited a significantly larger number (83% and 69%, respectively) of normal kits (in two litters, all 19 kits were normal); only 9% (3/34) of the kits from saline-treated dams were born normal.
Melanoma
Using a human melanoma xenograft mouse model, our nNOS-selective inhibitors inhibited melanoma. The immune system produces T-cells to fight infection and cancer, but cancers use the PD-1 pathway to avoid detection by T-cells, thereby causing immune tolerance. Blocking the nNOS-mediated signaling pathway with our nNOS-selective inhibitors diminishes PD-L1 expression (the protein on T-cells that activates PD-1, which inhibits T-cell proliferation) and inhibits the growth of melanoma. Our inhibitors act as small molecule checkpoint immunotherapeutics.
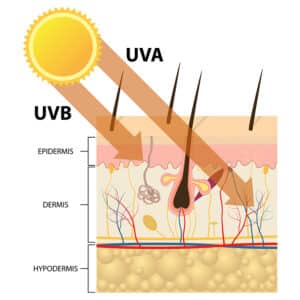 It has been well-documented that UV radiation, a major factor involved in melanoma genesis and disease progression, causes human skin to exhibit a remarkable increase in nitric oxide. nNOS is the enzyme that produces nitric oxide primarily in neuronal tissue, and as melanocytes originate from the neural crest and have many gene expression characteristics similar to neural cells, nNOS plays a prominent role in regulating nitric oxide levels in melanocytes.
It has been well-documented that UV radiation, a major factor involved in melanoma genesis and disease progression, causes human skin to exhibit a remarkable increase in nitric oxide. nNOS is the enzyme that produces nitric oxide primarily in neuronal tissue, and as melanocytes originate from the neural crest and have many gene expression characteristics similar to neural cells, nNOS plays a prominent role in regulating nitric oxide levels in melanocytes.
Patient biopsies have shown that compared to normal skin, malignant melanomas exhibit markedly higher expression levels of nNOS, which is significantly correlated with the disease stage.
Interferon-gamma (IFN-γ) exhibits profound stimulation of T cell immunity. The immune system produces T-cells to fight infection and cancer, but cancers can use the PD-1 pathway to avoid detection by T-cells. Preclinical studies demonstrate an enhancement of melanoma metastasis potential with IFN-γ exposure; an earlier clinical trial in melanoma patients also found that IFN-γ stimulates disease progression. Specific antibodies blocking IFN-γ, but not IFN-α, abolished UVB-induced melanocyte activation and melanoma development. IFN-γ also markedly induces the expression levels of nNOS in melanoma cells resulting in increased intracellular nitric oxide levels and increased melanoma.

Treatment with our novel nNOS inhibitors effectively alleviate IFN-γ-activation of transcription factors STAT1 and STAT3, which are important for cancer proliferation and chemotherapy resistance.
IFN-γ induces the expression of genes associated with melanoma proliferation, invasion, and immunosuppression, including programmed death-ligand 1 (PD-L1), a protein that activates programmed death receptor PD-1, the target of monoclonal antibody checkpoint inhibitors.
Blocking the nNOS-mediated signaling pathway with our nNOS-selective inhibitors effectively diminish IFN-γ-induced PD-L1 expression in melanoma cells. Using a human melanoma xenograft mouse model, it was found that IFN-γ increases tumor growth compared to control, which was inhibited by the co-administration of our nNOS inhibitors. Targeting nNOS with our inhibitors not only inhibits melanoma progression by blocking excessive NO production, but also diminishes IFN-γ-induced melanoma progression by inhibition of STAT1 and STAT3 activation and induction of PD-L1 expression. Given their mechanism to inhibit induction of PD-L1, they are small molecule checkpoint immunotherapeutics.